publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2025
-
 A unified pre-trained deep learning framework for cross-task reaction performance prediction and synthesis planningLi-Cheng Xu, Miao-Jiong Tang, Junyi An, and 2 more authorsNature Machine Intelligence, Sep 2025
A unified pre-trained deep learning framework for cross-task reaction performance prediction and synthesis planningLi-Cheng Xu, Miao-Jiong Tang, Junyi An, and 2 more authorsNature Machine Intelligence, Sep 2025Artificial intelligence has transformed the field of precise organic synthesis. Data-driven methods, including machine learning and deep learning, have shown great promise in predicting reaction performance and synthesis planning. However, the inherent methodological divergence between numerical regression-driven reaction performance prediction and sequence generation-based synthesis planning creates formidable challenges in constructing a unified deep learning architecture. Here we present RXNGraphormer, a framework to jointly address these tasks through a unified pre-training approach. By synergizing graph neural networks for intramolecular pattern recognition with Transformer-based models for intermolecular interaction modelling, and training on 13 million reactions via a carefully designed strategy, RXNGraphormer achieves state-of-the-art performance across eight benchmark datasets for reactivity or selectivity prediction and forward-synthesis or retrosynthesis planning, as well as three external realistic datasets for reactivity and selectivity prediction. Notably, the model generates chemically meaningful embeddings that spontaneously cluster reactions by type without explicit supervision. This work bridges the critical gap between performance prediction and synthesis planning tasks in chemical AI, offering a versatile tool for accurate reaction prediction and synthesis design.
-
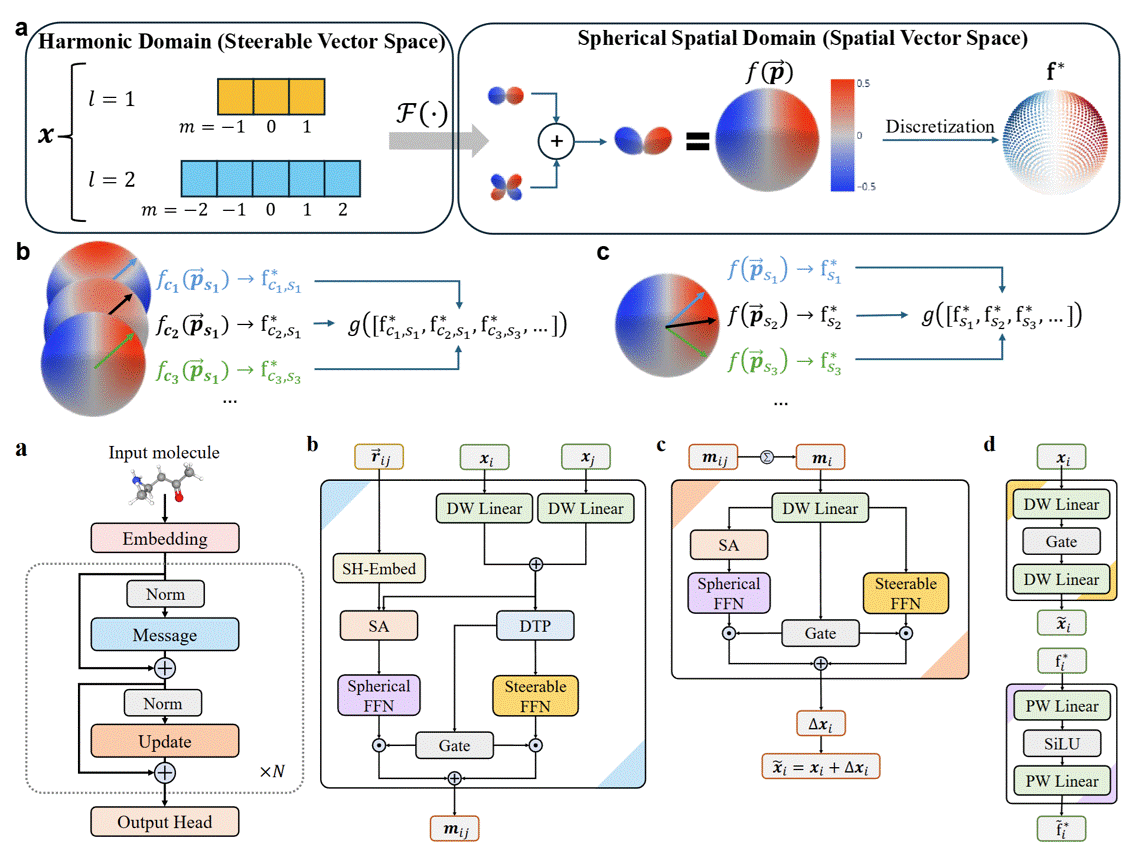 Equivariant Spherical Transformer for Efficient Molecular ModelingJunyi An, Xinyu Lu, Chao Qu, and 6 more authorsMay 2025arXiv:2505.23086 [cs]
Equivariant Spherical Transformer for Efficient Molecular ModelingJunyi An, Xinyu Lu, Chao Qu, and 6 more authorsMay 2025arXiv:2505.23086 [cs]SE(3)-equivariant Graph Neural Networks (GNNs) have significantly advanced molecular system modeling by employing group representations. However, their message passing processes, which rely on tensor product-based convolutions, are limited by insufficient non-linearity and incomplete group representations, thereby restricting expressiveness. To overcome these limitations, we introduce the Equivariant Spherical Transformer (EST), a novel framework that leverages a Transformer structure within the spatial domain of group representations after Fourier transform. We theoretically and empirically demonstrate that EST can encompass the function space of tensor products while achieving superior expressiveness. Furthermore, EST’s equivariant inductive bias is guaranteed through a uniform sampling strategy for the Fourier transform. Our experiments demonstrate state-of-the-art performance by EST on various molecular benchmarks, including OC20 and QM9.
-
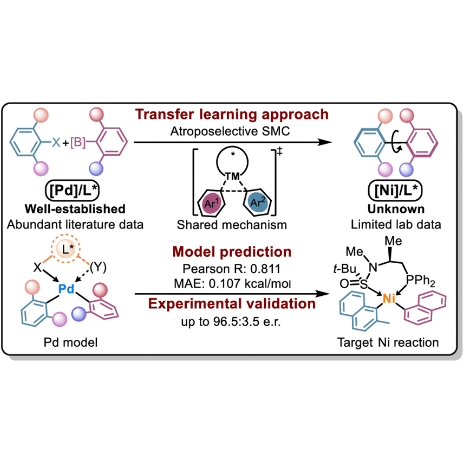 Transfer Learning-Enabled Ligand Prediction for Ni-Catalyzed Atroposelective Suzuki–Miyaura Cross-Coupling Based on Mechanistic Similarity: Leveraging Pd Knowledge for Ni DiscoveryXin-Yuan Xu, Li-Gao Liu, Li-Cheng Xu, and 2 more authorsJournal of the American Chemical Society, May 2025
Transfer Learning-Enabled Ligand Prediction for Ni-Catalyzed Atroposelective Suzuki–Miyaura Cross-Coupling Based on Mechanistic Similarity: Leveraging Pd Knowledge for Ni DiscoveryXin-Yuan Xu, Li-Gao Liu, Li-Cheng Xu, and 2 more authorsJournal of the American Chemical Society, May 2025The rational design of novel molecular catalysts often confronts challenges due to complex structure–performance relationships. Emerging data-driven approaches provide revolutionary solutions, yet the application of machine learning to new catalyst development inevitably faces a low-data regime with limited effective structure–performance modelings available. In this study, we present a transfer learning strategy to facilitate knowledge transfer from well-documented Pd catalysis to a novel, underexplored Ni system. By synergistically modeling extensive Pd catalysis data with limited Ni/Sadphos data, our approach accurately predicted novel Sadphos ligands, enabling the first atroposelective Ni-catalyzed Suzuki–Miyaura cross-coupling reaction. The synthetic utility of the machine learning-predicted ligand was further demonstrated in its broad synthetic scope, gram-scale synthesis, and precise control of dual axial chiralities in ternaphthalene through the sequential coupling under Ni and Pd catalysis. Additionally, density functional theory calculations were employed to reveal the reaction mechanism and stereochemical model of this new Ni catalyst, validating the proposed mechanistic connection between Ni and Pd. This work demonstrates how machine learning models can effectively leverage mechanistic connectivity, applying extensive structure–performance relationship data from the literature to predict new catalysts, providing a novel strategy for the rational design of molecular catalysts from a few-shot learning perspective.
2023
-
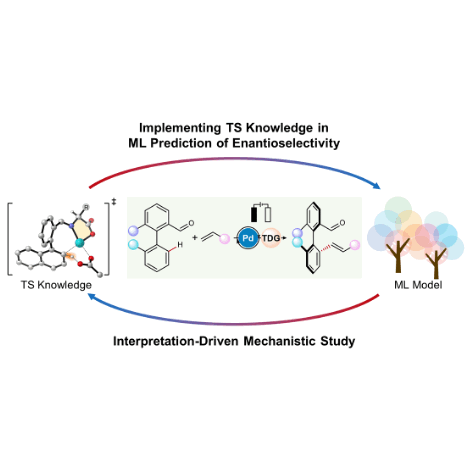 Enantioselectivity prediction of pallada-electrocatalysed C–H activation using transition state knowledge in machine learningLi-Cheng Xu, Johanna Frey, Xiaoyan Hou, and 6 more authorsNature Synthesis, Jan 2023
Enantioselectivity prediction of pallada-electrocatalysed C–H activation using transition state knowledge in machine learningLi-Cheng Xu, Johanna Frey, Xiaoyan Hou, and 6 more authorsNature Synthesis, Jan 2023Enantioselectivity prediction in asymmetric catalysis has been a long-standing challenge in synthetic chemistry because of the high-dimensional nature of the structure–enantioselectivity relationship. A lack of understanding of the synthetic space results in laborious and time-consuming efforts in the discovery of asymmetric reactions, even if the same transformation has already been optimized on model substrates. Here we present a data-driven workflow to achieve a holistic enantioselectivity prediction of asymmetric pallada-electrocatalysed C–H activation by implementing transition state knowledge in machine learning. The vectorization of transition state knowledge allowed for an excellent description and extrapolation of the machine learning model, and enabled the quantitative evaluation of 846,720 possibilities. Model interpretation revealed the non-intuitive olefin effect on the enantioselectivity determination. Subsequent density functional theory calculations unravelled mechanistic knowledge that the rate-determining step depends on the olefin reactivity in the insertion step. Therefore, the olefin insertion step can be involved in the overall enantioselectivity determination. These results highlight the complementary features of knowledge-based machine learning with an interpretation-driven mechanistic study, which provides the opportunity to harness widely existing catalysis screening data and transition state models in molecular synthesis.
-
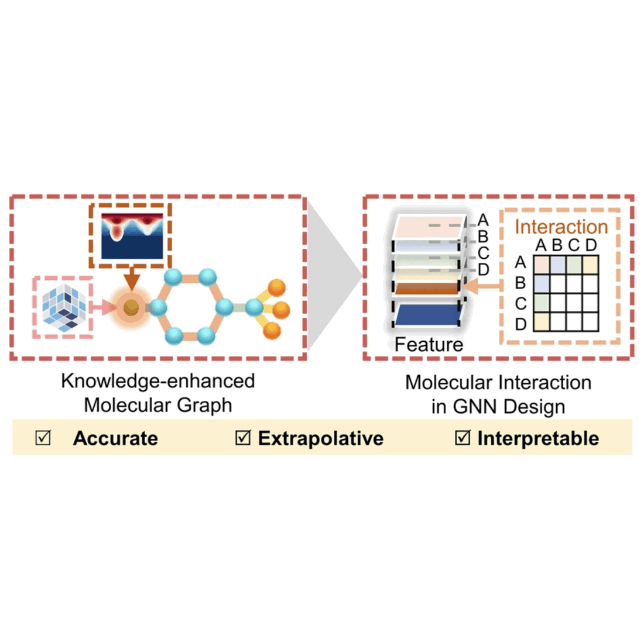 Reaction performance prediction with an extrapolative and interpretable graph model based on chemical knowledgeShu-Wen Li, Li-Cheng Xu, Cheng Zhang, and 2 more authorsNature Communications, Jun 2023
Reaction performance prediction with an extrapolative and interpretable graph model based on chemical knowledgeShu-Wen Li, Li-Cheng Xu, Cheng Zhang, and 2 more authorsNature Communications, Jun 2023Accurate prediction of reactivity and selectivity provides the desired guideline for synthetic development. Due to the high-dimensional relationship between molecular structure and synthetic function, it is challenging to achieve the predictive modelling of synthetic transformation with the required extrapolative ability and chemical interpretability. To meet the gap between the rich domain knowledge of chemistry and the advanced molecular graph model, herein we report a knowledge-based graph model that embeds the digitalized steric and electronic information. In addition, a molecular interaction module is developed to enable the learning of the synergistic influence of reaction components. In this study, we demonstrate that this knowledge-based graph model achieves excellent predictions of reaction yield and stereoselectivity, whose extrapolative ability is corroborated by additional scaffold-based data splittings and experimental verifications with new catalysts. Because of the embedding of local environment, the model allows the atomic level of interpretation of the steric and electronic influence on the overall synthetic performance, which serves as a useful guide for the molecular engineering towards the target synthetic function. This model offers an extrapolative and interpretable approach for reaction performance prediction, pointing out the importance of chemical knowledge-constrained reaction modelling for synthetic purpose.
-
 Bridging Chemical Knowledge and Machine Learning for Performance Prediction of Organic SynthesisShuo-Qing Zhang, Li-Cheng Xu, Shu-Wen Li, and 4 more authorsChemistry – A European Journal, Jan 2023
Bridging Chemical Knowledge and Machine Learning for Performance Prediction of Organic SynthesisShuo-Qing Zhang, Li-Cheng Xu, Shu-Wen Li, and 4 more authorsChemistry – A European Journal, Jan 2023Recent years have witnessed a boom of machine learning (ML) applications in chemistry, which reveals the potential of data-driven prediction of synthesis performance. Digitalization and ML modelling are the key strategies to fully exploit the unique potential within the synergistic interplay between experimental data and the robust prediction of performance and selectivity. A series of exciting studies have demonstrated the importance of chemical knowledge implementation in ML, which improves the model’s capability for making predictions that are challenging and often go beyond the abilities of human beings. This Minireview summarizes the cutting-edge embedding techniques and model designs in synthetic performance prediction, elaborating how chemical knowledge can be incorporated into machine learning until June 2022. By merging organic synthesis tactics and chemical informatics, we hope this Review can provide a guide map and intrigue chemists to revisit the digitalization and computerization of organic chemistry principles.
-
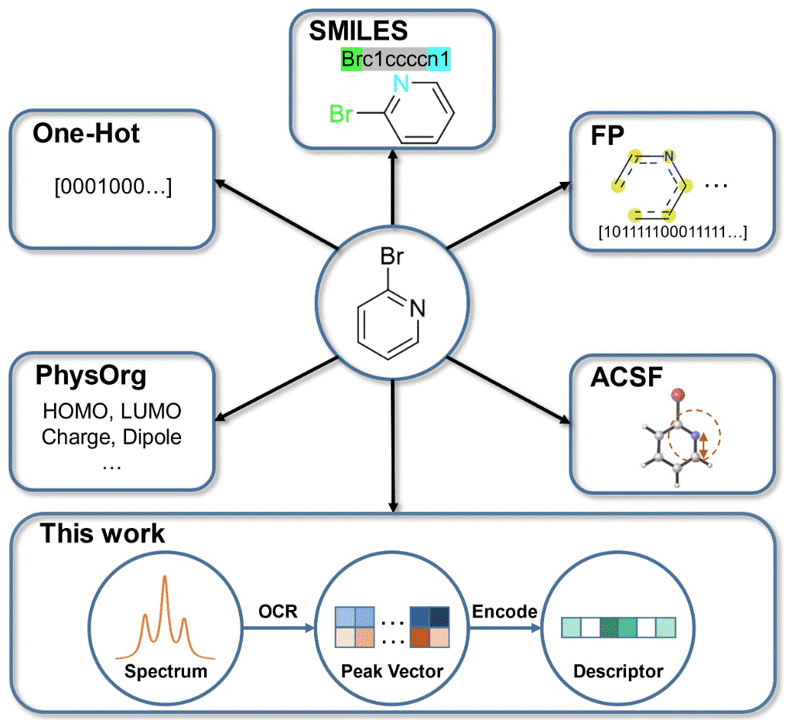 Exploring Spectrum-based Molecular Descriptors for Reaction Performance PredictionMiao-Jiong Tang, Li-Cheng Xu, Shuo-Qing Zhang, and 1 more authorChemistry – An Asian Journal, Apr 2023
Exploring Spectrum-based Molecular Descriptors for Reaction Performance PredictionMiao-Jiong Tang, Li-Cheng Xu, Shuo-Qing Zhang, and 1 more authorChemistry – An Asian Journal, Apr 2023Despite the availability and accuracy of modern spectroscopic characterization, the utilization of spectral information in chemical machine learning is still primitive. Here, we report an optical character recognition-based automatic process to utilize spectral information as molecular descriptors, which directly transforms experimental spectrum images to readable vectors. We demonstrate its machine learning application in the reaction yield dataset of Pd-catalyzed Buchwald-Hartwig cross-coupling with aryl halides. In addition, we also show that the predicted spectrum can serve as an alternative encoding source to support the model training.
-
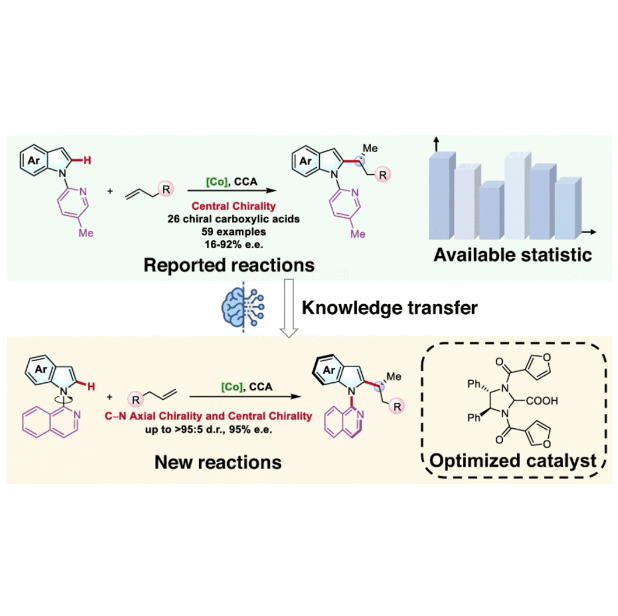 Data-driven design of new chiral carboxylic acid for construction of indoles with C-central and C–N axial chirality via cobalt catalysisZi-Jing Zhang, Shu-Wen Li, João C. A. Oliveira, and 7 more authorsNature Communications, May 2023
Data-driven design of new chiral carboxylic acid for construction of indoles with C-central and C–N axial chirality via cobalt catalysisZi-Jing Zhang, Shu-Wen Li, João C. A. Oliveira, and 7 more authorsNature Communications, May 2023Challenging enantio- and diastereoselective cobalt-catalyzed C–H alkylation has been realized by an innovative data-driven knowledge transfer strategy. Harnessing the statistics of a related transformation as the knowledge source, the designed machine learning (ML) model took advantage of delta learning and enabled accurate and extrapolative enantioselectivity predictions. Powered by the knowledge transfer model, the virtual screening of a broad scope of 360 chiral carboxylic acids led to the discovery of a new catalyst featuring an intriguing furyl moiety. Further experiments verified that the predicted chiral carboxylic acid can achieve excellent stereochemical control for the target C–H alkylation, which supported the expedient synthesis for a large library of substituted indoles with C-central and C–N axial chirality. The reported machine learning approach provides a powerful data engine to accelerate the discovery of molecular catalysis by harnessing the hidden value of the available structure-performance statistics.
-
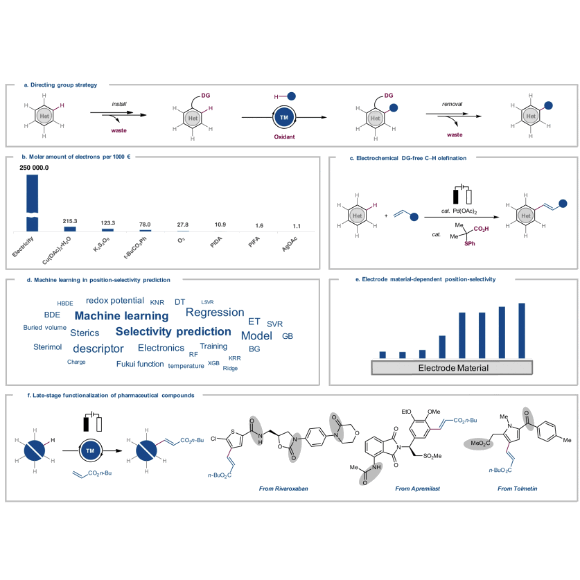 Electrocatalyzed direct arene alkenylations without directing groups for selective late-stage drug diversificationZhipeng Lin, Uttam Dhawa, Xiaoyan Hou, and 9 more authorsNature Communications, Jul 2023
Electrocatalyzed direct arene alkenylations without directing groups for selective late-stage drug diversificationZhipeng Lin, Uttam Dhawa, Xiaoyan Hou, and 9 more authorsNature Communications, Jul 2023Electrooxidation has emerged as an increasingly viable platform in molecular syntheses that can avoid stoichiometric chemical redox agents. Despite major progress in electrochemical C−H activations, these arene functionalizations generally require directing groups to enable the C−H activation. The installation and removal of these directing groups call for additional synthesis steps, which jeopardizes the inherent efficacy of the electrochemical C−H activation approach, leading to undesired waste with reduced step and atom economy. In sharp contrast, herein we present palladium-electrochemical C−H olefinations of simple arenes devoid of exogenous directing groups. The robust electrocatalysis protocol proved amenable to a wide range of both electron-rich and electron-deficient arenes under exceedingly mild reaction conditions, avoiding chemical oxidants. This study points to an interesting approach of two electrochemical transformations for the success of outstanding levels of position-selectivities in direct olefinations of electron-rich anisoles. A physical organic parameter-based machine learning model was developed to predict position-selectivity in electrochemical C−H olefinations. Furthermore, late-stage functionalizations set the stage for the direct C−H olefinations of structurally complex pharmaceutically relevant compounds, thereby avoiding protection and directing group manipulations.
2022
-
 When machine learning meets molecular synthesisJoão C.A. Oliveira, Johanna Frey, Shuo-Qing Zhang, and 5 more authorsTrends in Chemistry, Oct 2022
When machine learning meets molecular synthesisJoão C.A. Oliveira, Johanna Frey, Shuo-Qing Zhang, and 5 more authorsTrends in Chemistry, Oct 2022The recent synergy of machine learning (ML) with molecular synthesis has emerged as an increasingly powerful platform in organic synthesis and catalysis. This merger has set the stage for key advances in inter alia reaction optimization and discovery as well as in synthesis planning. The creation of predictive ML models relies on chemical databases, molecular descriptors, and the choice of the ML algorithms. Chemical databases provide a crucial support of chemical knowledge contributing to the development of an accurate and generalizable ML model. Molecular descriptors translate the chemical structure into digital language, so that substrates or catalysts in molecular synthesis and catalysis are represented in a numerical fashion. ML algorithms achieve an effective mapping between the molecular descriptors and the target properties, enabling an efficient prediction based on readily available or calculated descriptors. Herein, we highlight the key concepts and approaches in ML and their major potential towards molecular synthesis with emphasis in catalysis, pointing out additionally the most successful cases in the field.
2021
-
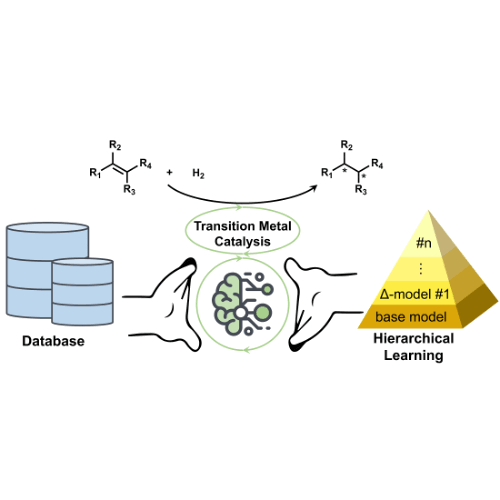 Towards Data-Driven Design of Asymmetric Hydrogenation of Olefins: Database and Hierarchical LearningLi-Cheng Xu, Shuo-Qing Zhang, Xin Li, and 3 more authorsAngewandte Chemie International Edition, Oct 2021
Towards Data-Driven Design of Asymmetric Hydrogenation of Olefins: Database and Hierarchical LearningLi-Cheng Xu, Shuo-Qing Zhang, Xin Li, and 3 more authorsAngewandte Chemie International Edition, Oct 2021Asymmetric hydrogenation of olefins is one of the most powerful asymmetric transformations in molecular synthesis. Although several privileged catalyst scaffolds are available, the catalyst development for asymmetric hydrogenation is still a time- and resource-consuming process due to the lack of predictive catalyst design strategy. Targeting the data-driven design of asymmetric catalysis, we herein report the development of a standardized database that contains the detailed information of over 12000 literature asymmetric hydrogenations of olefins. This database provides a valuable platform for the machine learning applications in asymmetric catalysis. Based on this database, we developed a hierarchical learning approach to achieve predictive machine leaning model using only dozens of enantioselectivity data with the target olefin, which offers a useful solution for the few-shot learning problem and will facilitate the reaction optimization with new olefin substrate in catalysis screening.
-
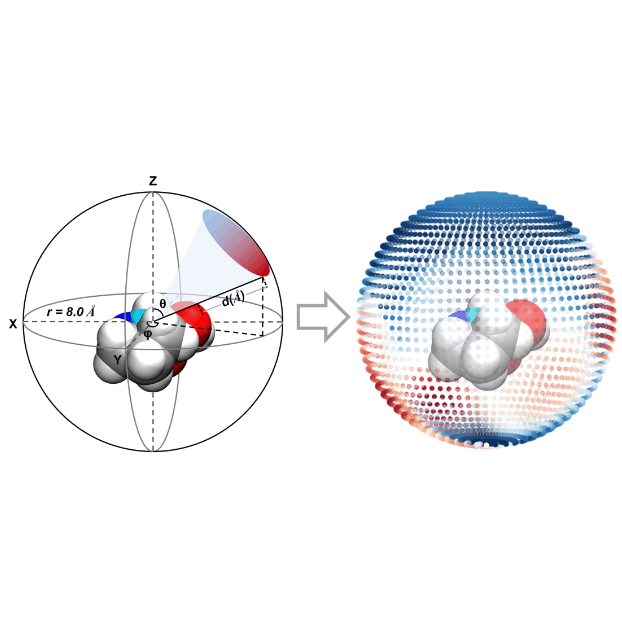 A Molecular Stereostructure Descriptor Based On Spherical ProjectionLi-Cheng Xu, Xin Li, Miao-Jiong Tang, and 4 more authorsSynlett, Nov 2021
A Molecular Stereostructure Descriptor Based On Spherical ProjectionLi-Cheng Xu, Xin Li, Miao-Jiong Tang, and 4 more authorsSynlett, Nov 2021Description of molecular stereostructure is critical for the machine learning prediction of asymmetric catalysis. Herein we report a spherical projection descriptor of molecular stereostructure (SPMS), which allows precise representation of the molecular van der Waals (vdW) surface. The key features of SPMS descriptor are presented using the examples of chiral phosphoric acid, and the machine learning application is demonstrated in Denmark’s dataset of asymmetric thiol addition to N-acylimines. In addition, SPMS descriptor also offers a color-coded diagram that provides straightforward chemical interpretation of the steric environment.
2020
-
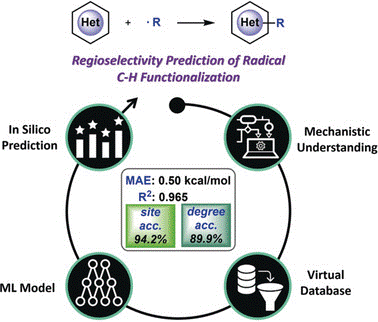 Predicting Regioselectivity in Radical C−H Functionalization of Heterocycles through Machine LearningXin Li, Shuo-Qing Zhang, Li-Cheng Xu, and 1 more authorAngewandte Chemie International Edition, Aug 2020
Predicting Regioselectivity in Radical C−H Functionalization of Heterocycles through Machine LearningXin Li, Shuo-Qing Zhang, Li-Cheng Xu, and 1 more authorAngewandte Chemie International Edition, Aug 2020Radical C−H bond functionalization provides a versatile approach for elaborating heterocyclic compounds. The synthetic design of this transformation relies heavily on the knowledge of regioselectivity, while a quantified and efficient regioselectivity prediction approach is still elusive. Herein, we report the feasibility of using a machine learning model to predict the transition state barrier from the computed properties of isolated reactants. This enables rapid and reliable regioselectivity prediction for radical C−H bond functionalization of heterocycles. The Random Forest model with physical organic features achieved 94.2 % site accuracy and 89.9 % selectivity accuracy in the out-of-sample test set. The prediction performance was further validated by comparing the machine learning results with additional substituents, heteroarene scaffolds and experimental observations. This work revealed that the combination of mechanism-based computational statistics and machine learning model can serve as a useful strategy for selectivity prediction of organic transformations.